Os pacientes com transtornos de coagulação sanguínea constituem um grupo que requer atenção e cuidados especiais na prática odontológica. A hemofilia e a doença de von Willebrand são as mais comuns das coagulopatias hereditárias e devem ser detectadas antes que qualquer tratamento odontológico seja realizado. Uma boa anamnese, associada a um bom exame físico e bucal podem ajudar na detecção dessas patologias.
O cirurgião-dentista deve estar preparado para oferecer o tratamento adequado a esses pacientes, o qual depende da severidade da doença e do tipo de procedimento a ser realizado. Procedimentos menos invasivos geralmente não necessitam de cuidados especiais e podem ser realizados rotineiramente, desde que alguns cuidados básicos sejam tomados.
Cirurgias e técnicas anestésicas de bloqueio do nervo alveolar inferior devem ser realizadas com maior precaução, a fim de minimizar os riscos de sangramento e outras complicações. O hematologista deve ser consultado sempre que qualquer tratamento invasivo for planejado.
Vejamos o que a filologia tem a nos dizer acerca de alguns termos que aparecerão com frequência neste trabalho:
a.Diátese - di.á.te.se – é substantivo feminino e origina-se do grego “diáthesis”. Em medicina refere-se a condição do organismo para ser atacado por determinadas doenças ou estado mórbido geral manifestado por elas. [ ... ].(Dicionário Michaelis).
b.Hemofilia - he.mo.fi.li.a – é substantivo feminino e origina-se do grego “Haima”, ‘sangue’ e “Philia”, ‘gostar’. (não foi um neologismo muito feliz, como se vê). Em medicina refere-se a uma diátese congênita hereditária para hemorragias profusas e dificilmente controláveis; espontâneas ou causadas pelos menores traumatismos, em virtude de uma extrema fragilidade das paredes vasculares, e insuficiente e muito lenta coagulabilidade do sangue. Manifesta-se somente no sexo masculino. [ ... ]. (Dicionário Michaelis).
c.Coagular – verbo -vem o latim “coagulu” + “-are”. [ ... ];Transformar(-se) um líquido em matéria mais sólida. = coalhar, solidificar; [ ... ]. (Dicionário Priberam da Língua Portuguesa).
d.Coágulo -co.á.gu.lo – é substantivo masculino e vem o latim “coagulu” significando a parte coagulada ou coalhada de um líquido; coalho. (Dicionário Michaelis).
e.Coagulopatia – vem do latim “coágulu” + “-patia” é substantivo feminino que em medicina significa perturbação patológica da coagulação de sangue devida a transtorno ou carência de fatores plasmáticos que intervêm na coagulação. (Infopédia).
COAGULAÇÃO SANGUÍNEA
Recorrendo novamente à filologia encontramos que a palavra ‘hemostase’ é substantivo feminino e vem do grego “hemo+estase” significando, na acepção médica: 1 Estagnação de sangue em um vaso ou parte do corpo.2 Estancamento de uma hemorragia por meios médicos ou cirúrgicos.
A dificuldade em se obter uma boa hemostasia (estancamento do sangramento) pode ocorrer durante a prática odontológica, sendo assim importante que o cirurgião dentista tenha conhecimento de alguns recursos terapêuticos disponíveis.
Dentre eles, vem se mostrando bastante eficiente os adesivos fibrínicos, pois além de controlar a hemorragia também promovem adesão dos tecidos substituindo o procedimento de sutura sendo, deste modo, muito utilizados em situações emergenciais ou no tratamento de pacientes com distúrbios de coagulação.
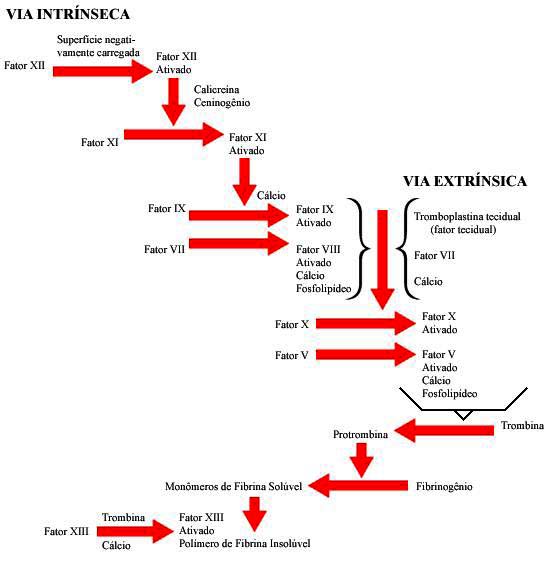
Como já sabemos o mecanismo da coagulação tem fundamental importância para obtenção da hemostasia, nele são descritos o que conhecemos como efeito cascata, onde a partir de uma lesão vascular, ocorre uma série de reações que objetivam a formação de uma rede de fibrina, que é a principal responsável pela manutenção da hemostasia. Este mecanismo pode ser dividido em fases (ROBERT et al.,1991) e será relembrado de forma resumida.
A primeira fase apresenta duas vias no processo de coagulação, chamadas de via intrínseca e via extrínseca (siga o texto seguinte com base na figura abaixo).
A via extrínseca é iniciada com lesão do tecido e liberação de tromboplastina tecidual. Nesta via a formação do ativador extrínseco se dá quando o fator III (lipoproteína) é liberado pelos tecidos que estão em torno do vaso sanguíneo, de modo que a fração proteica deste fator ativa o fator VII na presença de cálcio e o fator X ativa o fator V que vai agir com a fração lipídica do fator III.
A via intrínseca é iniciada pela exposição do sangue a uma superfície negativamente carregada. Nesta via a formação do ativador intrínseco se dá quando o fator XII se ativa espontaneamente no momento em que entra em contato com as bordas da lesão vascular.
Uma vez ativado ele dá origem a uma reação em cascata que consiste na ativação do fator XI na presença do cálcio. O fator XI ativará o fator IX que ativará o fator VIII que ativará o fator X que ativará o fator V. Esse fator V reagirá com fosfolipídios liberados pelas plaquetas, originando desta forma o ativador intrínseco.
Numa Segunda fase ocorrerá a transformação da protrombina em trombina, transformação esta catalisada pelos ativadores extrínsecos e intrínsecos formados na fase anterior.
Posteriormente, dentro do que ROBERT et al. (1991), chama de terceira fase do processo de hemostasia, a protrombina catalisa a transformação de fibrinogênio em fibrina.
Numa próxima etapa (quarta fase), ocorrerá a polimerização das moléculas de fibrina transformando-as em filamentos que se entrelaçam formando a rede de fibrina, responsável pela retenção do sangue em suas malhas, originando assim o coágulo sanguíneo. Esta fase é catalisada pelo fator XIII e o coágulo formado é uma estrutura frouxa que pode ser deslocado pela própria pressão do sangue .
Na quinta fase ocorre a retração do coágulo, que se caracteriza pela expulsão da água e dos sais minerais, retendo apenas a parte celular e as proteínas plasmáticas, assim o coágulo passa a apresentar uma estrutura mais sólida e resistente.
A última fase (sexta fase) é caracterizada pela fibrinólise, que consiste na reabsorção gradual do coágulo simultaneamente com a cicatrização da parede do vaso sanguíneo, processo este que ocorre 24 a 48 horas após o início da coagulação, de forma que a substância responsável pela reabsorção do coágulo é a fibrinolisina, substância esta ativada pelo fator XIII.
COAGULOPATIAS
As coagulopatias podem ser genéticas ou adquiridas. As mais frequentes são as hereditárias e este trabalho vai tratar fundamentalmente delas.
COAGULOPATIAS HEREDITÁRIAS
São doenças hemorrágicas resultantes da deficiência quantitativa e/ou qualitativa de uma ou mais das proteínas plasmáticas (fatores) da coagulação e são normalmente representadas pelas hemofilias A e B e pela Doença von Willebrand.
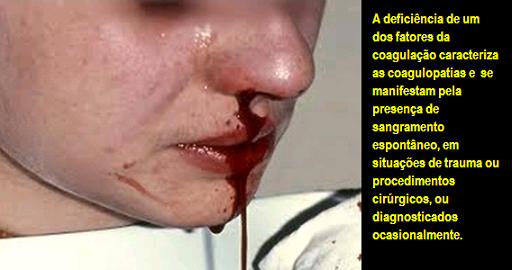
A maioria dos pacientes apresenta deficiência quantitativa ou qualitativa de um dos fatores da coagulação (fator VIII para a hemofilia A ou fator IX para o B). A deficiência de um desses fatores se manifesta pela presença de sangramento espontâneo, em situações de trauma ou procedimentos cirúrgicos, ou diagnosticados ocasionalmente.
Sob um critério estritamente etiológico, as coagulopatias hereditárias podem ser classificadas de acordo com as seguintes deficiências de fator:
1.Deficiência de fibrinogênio (fator I), que se subdivide em:
1.1.Afibrinogenemia
1.2.Hipofibrinogenemia
1.3.Disfibrinogenemia
2.Deficiência de protrombina (fator II)
3.Deficiência de fator V
4.Deficiência de fator VII
5.Deficiência de fator VIII - Hemofilia A
6.Deficiência de fator IX - Hemofilia B
7.Deficiência de fator X
8.Deficiência de fator XI
9.Deficiência de fator XII
10.Deficiência do fator estabilizador da fibrina (fator XIII)
11.Doença von Willebrand
Dentre as coagulopatias hereditárias, as hemofilias A e B e a doença von Willebrand (DVW) são as mais comuns. São consideradas coagulopatias raras as deficiências de fatores I, II, V, VII, X e XIII.
HEMOFILIAS A e B
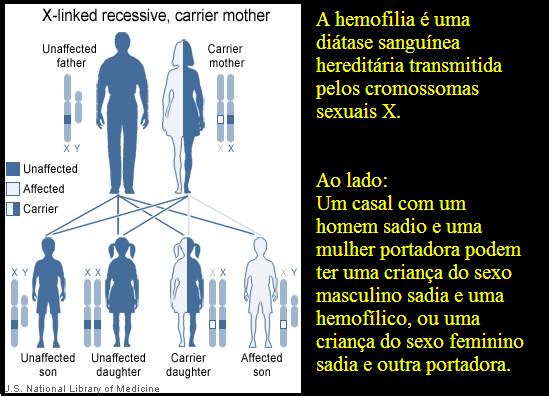
A hemofilia é uma doença hereditária hemorrágica, ligada ao cromossoma X.
As manifestações hemorrágicas no primeiro ano de vida são observadas nas formas moderadas e graves, sendo os hematomas secundários a injeções intramuscular (IM), punções venosas, traumas e sangramentos em mucosa oral as mais frequentes.
Raramente, observa-se no recém-nascido hemorragia no coto umbilical e no sistema nervoso central (SNC). Como sangramentos no SNC podem ocorrer, muitas vezes espontaneamente, cefaléias não explicadas devem ser tratadas como hemorragias intracranianas, até que o quadro se esclareça.
Outra hemorragia potencialmente grave é a retroperitonial (localizada atrás do peritôneo), que pode ser volumosa a ponto de causar choque hipovolêmico (diminuição acentuada do volume sanguíneo), se tratada tardiamente.
DOENÇA DE VON WILLEBRAND
É uma doença hemorrágica hereditária causada por uma diminuição ou uma disfunção da proteína chamada fator de von Willebrand (FvW).
Isto ocorre devido à mutação no cromossomo 12 e é caracterizada por deficiência qualitativa ou quantitativa do fator de von Willebrand. A diversidade de mutações leva ao aparecimento das mais variadas manifestações clínicas possibilitando a divisão dos pacientes em vários tipos e subtipos clínicos.
A coagulopatia se manifesta basicamente através da disfunção plaquetária associada à diminuição dos níveis séricos do fator coagulante VIII (o mesmo fator implicado na hemofilia A), existindo também casos raros de doença de von Willebrand adquirida.
Foi descrita pela primeira vez em 1925 pelo médico finlandês Erik Adolf von Willebrand. Essa é a doença hemorrágica mais comum e atinge cerca de 2% da população mundial, alcançando igualmente ambos os sexos; porém, mulheres têm mais probabilidade de ter a doença diagnosticada pelas manifestações durante a menstruação.
PERGUNTAS E RESPOSTAS
O que é hemofilia?
Normalmente, quando ocorre um sangramento, o organismo inicia uma resposta em duas etapas para interromper o episódio.
Na primeira, as plaquetas tornam-se viscosas e acumulam-se no local do ferimento formando um tampão. Depois, o sistema de coagulação transforma o tampão de plaquetas instável em um coágulo de fibrina quimicamente estável. Isso acontece por uma série de reações enzimáticas envolvendo substâncias chamadas fatores de coagulação.
Na hemofilia, há deficiência ou ausência dos fatores VIII ou IX da coagulação. Por isso, quando ocorre uma lesão no vaso sanguíneo, o processo de coagulação não ocorre de forma adequada, resultando em um sangramento descontrolado. Pessoas com falta de fator VIII têm hemofilia A e pessoas com insuficiência de fator IX têm hemofilia B.
Como se contrai a hemofilia?
Na maioria dos casos a hemofilia é hereditária e afeta principalmente os homens. As mulheres podem carregar o gene que causa a hemofilia. Quando uma mulher que é portadora têm filhos, há uma probabilidade de 50% que os meninos desenvolvam a doença e 50% de probabilidade que as meninas tornem-se portadoras.
Enquanto filhos de homens hemofílicos não herdam a doença, todas as filhas deles serão portadoras. Além disso, em cerca de um terço dos casos, não há história familiar da doença e hemofilia ocorrendo como resultado de uma nova mutação genética.
O que causa a doença? É verdade que a doença é mais prevalente em homens? Por que isso ocorre?
A hemofilia é herdada em condição recessiva ligada ao cromossomo X, acometendo quase que exclusivamente indivíduos do sexo masculino.
Pergunta: Como é o tratamento?
O tratamento é feito através da reposição do fator deficiente com concentrado do fator da coagulação específico, isto é, do fator VIII ou fator XI. Pode ser feito sob demanda ou profilático (preventivo). O tratamento de demanda deve ser instituído imediatamente na evidência de sangramento.
Em que casos a transfusão de sangue é recomendada?
Nos casos de sangramento intenso, quando houver anemia importante.
Que recomendações são importantes para que o hemofílico tenha uma melhor qualidade de vida?
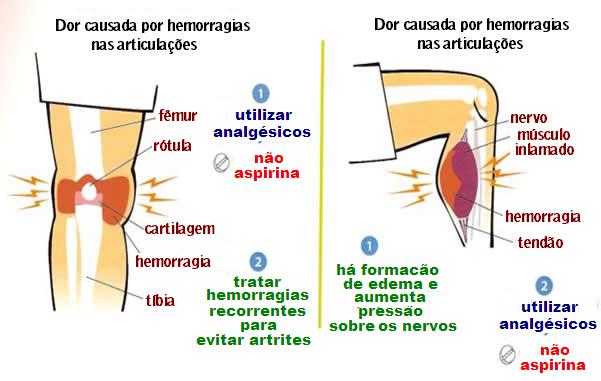
Conforme recomendação da Organização Mundial da Saúde/Federação Mundial da Hemofilia o principal objetivo é impedir o sangramento articular e suas sequelas, através da profilaxia, pois, a artropatia (doença nas articulações) crônica hemofílica é a maior causa de morbidade dos pacientes.
Quais são os sinais e sintomas da hemofilia?
A mais séria hemorragia é a associada à hemofilia tende a ser interna porque, diferentemente da hemorragia causada por ferimentos, pode ser invisível e difícil de antecipar.
A hemorragia na hemofilia pode ocorrer quase em qualquer lugar do corpo e pode danificar articulações, ossos, nervos e outros tecidos.
Os sintomas incluem contusões frequentes ou sangramentos repetidos nas articulações como joelhos, cotovelos e quadril. Algumas vezes, quando a hemorragia ocorre em músculos profundos ou outros tecidos moles, pode ser percebida como um músculo distendido.
Quando o sangramento ocorre no antebraço, panturrilha ou virilha, o intumescimento pode causar pressão sobre os nervos, resultando em dormência, dor e incapacidade de movimento do membro.
O atendimento médico imediato deve ser procurado nesses casos. A hemorragia pode também ocorrer na boca, face, pescoço ou garganta.
Isso pode ser sério porque há possibilidade de obstrução da respiração. Hemorragia dentro da cabeça, se causada por um ferimento ou sem uma razão aparente (hemorragia espontânea) pode não exibir sintomas por vários dias.
Sintomas a serem procurados incluem irritabilidade, sonolência, sensação de torpor ou formigamento, dor de cabeça, confusão, náusea, vômito, dificuldade de falar ou engolir e mudança na visão. Todos os ferimentos na cabeça são sérios e requerem atenção médica imediata.
m 10pt" class=MsoNormal>
Como a hemofilia afeta o estilo de vida?
A hemofilia é uma condição vitalícia. Embora atualmente não haja cura, existem terapias de controle da hemorragia que possibilitam aos hemofílicos a condução de uma vida relativamente normal.
Pais de crianças nessa condição devem ter as mesmas precauções que quaisquer pais e igualmente escolher brinquedos macios e roupas acolchoadas.
A socialização e as brincadeiras com outras crianças são importantes para o desenvolvimento da criança e devem ser encorajados. Quando a criança cresce, assuntos relacionados a esportes e namoro devem ser discutidos.
Os jovens podem querer trocar ideias sobre como a hereditariedade dessa doença pode afetar sexualidade, casamento e planejamento familiar.
No decorrer da vida, os exercícios físicos são importantes. Músculos mais fortes sustentam as articulações e podem reduzir o número de sangramentos. Deve-se consultar um médico para desenvolver um programa de exercícios regulares que podem incluir, dependendo de preferências pessoais, natação, caminhadas, e outras atividades que causem menor impacto nas articulações.
Como a hemofilia é tratada?
Como a hemofilia é resultado de deficiência ou ausência de certos fatores de coagulação, o tratamento envolve tipicamente a suplementação do fator ausente por administração intravenosa. A terapia de reposição do fator funciona para a maioria dos pacientes a menos que eles desenvolvam inibidores aos fatores de coagulação.
O que é um inibidor?
Devido ao fato de os fatores usados na terapia de reposição ser externos ao corpo, há sempre o risco do sistema imunológico do organismo reconhecê-los como estranhos e atacá-los.
Esse ataque é realizado pelos anticorpos. Normalmente, anticorpos ajudam a proteger o organismo da destruição de substâncias nocivas, e neutralizam os fatores VIII ou IX que foram adicionados ao sangue para parar a hemorragia (promover a hemostasia).
Qual teste é usado para achar inibidores?
Um tipo de teste conhecido como pesquisa e dosagem de inibidores é usado para verificar se a pessoa tem inibidores. Esse teste mede a concentração de anticorpos em unidades Bethesda. Quanto mais alto o número de unidades Bethesda, mais inibidores a pessoa tem.
Os inibidores são comuns?
Cientistas estimam que 15 a 20% das pessoas comhemofilia Asevera podem desenvolver inibidores em algum período de suas vidas. Os inibidores são muito menos comuns na hemofilia B, afetando apenas de 2 a 5% dos pacientes.
Quem está propenso a desenvolver inibidores?
Pessoas com risco mais elevado de adquirir inibidores são crianças (inibidores desenvolvem-se mais comumente antes dos 20 anos), pessoas com hemofilia grave e pessoas com história familiar de inibidores.
Cientistas estão estudando atualmente o gene da hemofilia e o sistema imunológico para aprender mais sobre a razão pela qual algumas pessoas desenvolvem inibidores mais frequentemente que outras.
Como as pessoas descobrem que têm inibidores?
Inibidores são detectados de duas maneiras:
1.Durante um episódio hemorrágico, quando o sangramento não para tão rapidamente quanto deveria depois de uma dose regular de fator VIII ou fator IX;
2.Durante uma visita ao consultório, por meio de um exame de sangue.
Qual teste é usado para achar inibidores?
Um tipo de teste conhecido como pesquisa e dosagem de inibidores é usado para verificar se a pessoa tem inibidores. Esse teste mede a concentração de anticorpos em unidades Bethesda. Quanto mais alto o número de unidades Bethesda, mais inibidores a pessoa tem.
Há mais de um tipo de inibidor?
Inibidores são classificados pela forma com a qual respondem ao tratamento dos fatores VIII ou IX. Eles são considerados como de “alta resposta” ou de “baixa resposta”.
Tipo de inibidor
Tipo de Resposta Imune
Reação ao uso repetido de fatores VIII e IX
Alta resposta
Forte
Níveis do inibidor aumentam rapidamente
Baixa resposta
Mais lenta e mais fraca
Níveis do inibidor pouco aumentam ou permanecem estáveis
O nível do inibidor pode modificar-se ao decorrer do tempo, mudando de baixo para alto ou de alto para baixo, conforme a resposta.
Pacientes cuja resposta é baixa podem ser tratados usualmente com uma dose mais alta de fatores VIII e IX; pacientes de alta resposta necessitam de outras alternativas.
Como os episódios de hemorragia são tratados em pessoas com inibidores?
Tratar pacientes com inibidores é desafiador porque cada paciente reage diferentemente aos tratamentos e produtos disponíveis. Recentemente, vários modos foram desenvolvidos para o tratamento de indivíduos com inibidores aos fatores VIII e IX. A tabela abaixo lista as várias terapias que têm sido usadas nos últimos 20 anos.
Terapias de Tratamento de Episódios de Hemorragia em Pacientes com Inibidores
Terapia
Quando da introdução
Como funciona
Concentrados de complexo Protrombínicos (CCPs)
1970 (década)
Contém vários outros fatores de coagulação derivados do plasma humano do processo de coagulação do que o fator VIII, evitando a necessidade do fator VIII para alcançar a coagulação
Concentrados de complexo protrombínicos parcialmente ativados (CCPa)
1970 (década)
Contém protrombina derivada do plasma humano ativada e outros fatores de coagulação, evitando portando a necessidade do fator VIII para alcançar a coagulação
Plasmaferese
1970 (década)
O sangue é processado e o inibidor é removido do plasma; então o plasma é devolvido ao paciente e o tratamento pode continuar com fator VIII ou IX ou outras alternativas
Terapia de imuno-tolerância
Final de 1970
Fator VIII é administrado em doses altas e freqüentes por um período que varia de semanas a anos; o resultado desejado é que os inibidores parem de ser produzidos pelo organismo de forma que o paciente possa responder aos fatores VIII e IX
Fator VIII porcino
1980
Fator VIII, derivado do plasma suíno, é levemente diferente do fator VIII humano, podendo passar sem reconhecimento pelos inibidores do fator VIII. (Essa terapia está aprovada somente para pacientes com hemofilia A)
Fator VIIa Recombinante
1996
Evita os fatores VIII e IX no processo de coagulação; por ser recombinante, elimina o risco de contaminação viral humana que é sempre uma preocupação nos produtos derivados do plasma humano
UMA PEQUENA HISTÓRIA DA HEMOFILIA
Existe registro da hemofilia no texto judaico, o Talmud (cerca de 50-130dC). Segundo o Talmud uma criança não deveria ser circuncidada se já tivessem morrido dois irmãos em tal procedimento.
No século XII, o médico árabe Abulcasis descreveu um caso de uma família cujos homens haviam morrido de sangramento após pequenos ferimentos.
Em 1803, o médico norte-americano John Conrad constatou que havia "tendências a sangramentos em algumas famílias". Ele chegou a conclusão que a doença era hereditária e acometia mais homens do que mulheres.
O termo hemofilia apareceu pela primeira vez em 1828 por Hopff da Universidade de Zurique. Em 1937, Patek e Taylor, dois médicos de Harvard descobriram a globulina anti-hemofílica.
Pavlosky, um médico de Buenos Aires, separou a Hemofilia A e Hemofilia B laboratorialmente. Este teste era feito transferindo o sangue de um hemofílico para outro hemofílico. O fato corrigia o sangramento, comprovando que havia mais de um tipo de hemofilia.
A Hemofilia é, muitas vezes, associada à história da Monarquia na Europa. A rainha Vitória passou a doença ao seu filho Leopoldo, e através de várias das suas filhas, a várias famílias reais Europeias, incluindo as famílias reais da Espanha, Alemanha, e Rússia. Alexei Romanov, filho do Czar Nicolau II da Rússia, foi um dos descendentes da Rainha Vitória que herdou a doença.
Durante as décadas de 1970 e 1980 a falta de outras modalidades de tratamento e a insuficiência de tecnologia para diagnosticar alguns vírus como o da AIDS acarretou em contaminação em grande escala da população hemofílica que dependiam de constantes transfusões de sangue.
Mas este é um cenário que não está totalmente no passado, a maioria absoluta, em torno de 75%, da população mundial de hemofílicos não dispõe dos medicamentos básicos para o tratamento desta patologia, ficando ainda, estes, sujeitos às transfusões de sangue que pode lhes salvar a vida como também pode tirá-la.
CONDUTA ODONTOLÓGICA
INTRODUÇÃO:
Com o desenvolvimento dos concentrados de fatores da coagulação, especialmente os concentrados de alta pureza de Fator VIII e IX, bem como, os concentrados de fatores monoclonais e recombinantes, um gigantesco passo foi dado para a terapia de reposição nos pacientes com distúrbios hemorrágicos.
Infelizmente, este avanço não aconteceu no campo odontológico. É muito comum ver jovens e adultos hemofílicos com múltiplas cáries e sérios problemas periodontais, sem qualquer possibilidade de restauração, não só por falta de entendimento entre o dentista e o hematologista, como também, por medo dos próprios hemofílicos.
Fez-se, então, necessário criar clínicas dentárias em HEMOCENTROS visando solucionar estas carências. Por outro lado, instituiu-se, nestes locais, um atendimento dentário aos pacientes com discrasias sanguíneas, e, especialmente, aos hemofílicos e aos pacientes com doença de von Willebrand.
E desde, então, tem-se obtido um excelente resultado no tratamento dentário conservador e no trabalho de prevenção, junto às famílias destes pacientes.
PLANO DE TRATAMENTO ODONTOLÓGICO:
Para iniciarmos o tratamento em pacientes hemorrágicos é necessário um perfeito estudo dos seus problemas odontológicos. O seguinte plano deverá ser seguido:
1EXAME CLÍNICO
Como qualquer paciente, o hemofílico deve ser submetido a um perfeito exame odontológico, que é constituído de:
- anamnese e histórico do paciente;
- tipo de hemofilia e grau de severidade;
- presença de inibidor;
- história familiar e medicação usada no controle da dor.
O tratamento deve ser sempre documentado no prontuário, para as consultas com o médico hematologista, responsável pelo paciente, ou para discussão no GRUPO DE HEMOFILIA que, por ventura, o paciente frequenta.
As radiografias dentárias são indispensáveis, e, se possível, deve-se utilizar radiografias panorâmicas.
No caso de “modelos de estudo” serem feitos, as bordas das moldeiras devem ser protegidas com cera, a fim de serem mínimos os traumatismos nos tecidos moles da boca.
2PROFILAXIA E ADEQUAÇÃO BUCAL
É sempre feita na primeira seção. A remoção da placa bacteriana e do tártaro (cálculo) supra-gengival não ocasiona grande sangramento, entretanto, deve ser efetuada, cuidadosamente.
A gengivite, por exemplo, pode predispor o paciente a sangramento gengival espontâneo.
Nesta ocasião também pode ser feito o selamento de cavidades com o ionômero de vidro, polimento coronário e instruções de higiene.
Também, na primeira consulta, devem ser aparados e arredondados os restos radiculares ou os remanescentes coronários com bordos cortantes, que podem atuar como lâminas afiadas, principalmente no assoalho da língua.
Quando a curetagem for subgengival, ela deve ser efetuada com o paciente previamente transfundido de fatores da coagulação. Ela deve ser realizada com muito cuidado e somente em dois ou três elementos por sessão.
3 PREVENÇÃO DAS CÁRIES
- Higiene bucal – podemos obter uma grande redução do número de cáries dentárias e de problemas periodontais, se o paciente for instruído no sentido de ter uma ótima higiene bucal, mediante uma correta escovação e o uso de fio dental. Atuar coletivamente através de instruções para os pais e, se possível, com a projeção de filmes ou slides educativos, etc.
- Fluoretação - a aplicação tópica de flúor e os bochechos com soluções fluoretadas são importantes para as crianças.
- Aplicação de selantes.
- Nutrição e dieta - é necessária uma dieta balanceada e adequada, rica em vitaminas, sais minerais e controle na ingestão de sacarose. Os pais devem ser alertados sobre os perigos de formação da placa bacteriana.
4ANESTESIA
A anestesia, que é tema ainda de alguma controvérsia, é por nós largamente empregada.
Utilizamos a anestesia local, tanto na forma infiltrativa como regional ou troncular.
- A anestesia local ou infiltrativa pode ser ministrada em qualquer caso sem que se apresentem efeitos colaterais.
- A anestesia troncular ou regionaldeve ser evitada, pois expõe o paciente ao grande risco de ter um hematoma que pode bloquear as vias aéreas. Ela deve ser utilizada, somente, quando o paciente for previamente transfundido com o fator do qual ele tem deficiência.
5.TRATAMENTO DENTÁRIO CONSERVADOR
Este deve ser conduzido, rotineiramente, e deve ser feito como em qualquer outro paciente (exceto, se empregar a anestesia troncular). Neste caso, faz-se necessário o cuidado de afastar e proteger as partes moles (língua e bochechas), quando estivermos utilizando a broca de alta rotação, no preparo de cavidades, para receber obturações ou coroas.
Sempre que forem utilizadas matrizes de aço e as cunhas de madeira, elas devem ser colocadas com muito cuidado, para não lesar o tecido gengival.
5 TRATAMENTO ENDODÔNTICO (‘CANAL’)
É essa especialidade que recupera um dente, quer através da preservação da vitalidade pulpar, quer pelo tratamento do canal radicular, ou ainda pela recuperação da sua cor natural, cuja exodontia significaria a colocação de uma prótese de maior valor econômico. Portanto:
- não existe contraindicação para tratamento endodôntico em pacientes hemofílicos.
- nas pulpotomias e pulpectomias, a hemorragia pulpar não constitui problema podendo ser controlada por métodos usuais (epinefrina ou formocresol), mesmo em pacientes hemofílicos com inibidores.
- a anestesia troncular não é necessária, se a anestesia local for administrada com a técnica modificada.
- o tratamento endodôntico representa uma enorme economia, não só para o paciente, mas também para a instituição.
6TRATAMENTO PERIODONTAL
Os problemas periodontais são uma das maiores causas, entre adultos , da extração dentária. Portanto, os hemofílicos devem receber um excelente tratamento periodontal. O polimento coronário e a remoção de tártaros não ocasionam grande sangramento podendo ser feitos rotineiramente.
As gengivectomias, as gengivoplastias e os acessos para raspagem devem ser efetuados com o paciente previamente transfundido com o fator ausente.
7TRATAMENTO ORTODÔNTICO
Não existe nenhuma contraindicação para o tratamento ortodôntico; ao contrário, a correção de problemas oclusais e de estética será um grande benefício para o paciente.
Quando necessário, para evitar a laceração de mucosas e de lábios, deve-se proteger os fios e os ‘brackets’ com cera ou silicone protetor.
8PRÓTESES
Não existe contraindicação para “roachs” (prótese parcial removível – PPR), prótese fixa ou dentaduras parciais ou totais. Entretanto, as próteses devem ser confeccionadas por profissionais competentes.
9IMPLANTES
Os implantes dentários devem ser vistos com cautela, devido à complexidade do ato operatório.
10EXTRAÇÕES DENTÁRIAS
10.01. PRÉ-OPERATÓRIO:
- O paciente deve ser avaliado pelo hematologista e pelo dentista.
- Dosagem do fator, pesquisa de inibidores e raios X dentários devem ser verificados.
- Para as exodontias, recomenda-se (a critério do hematologista responsável), elevar a 30% o fator VIII (para hemofílicos A) e IX (para hemofílicos B), em dose única, repetindo se necessário.
- O uso de antifibrinolíticos deve, também ser avaliado.
- Antibióticos e tranquilizantes devem ser administrados sempre que forem necessários.
- Recomenda-se dieta líquido pastosa e fria.
- As extrações simples acarretam menor e menos possibilidade de sangramento, que são sempre proporcionais ao tamanho da ferida.
- As extrações múltiplas (intercaladas), no entanto, representam alguma vantagem: tempo de tratamento e economia do material de reposição.
- Quando avaliamos a radiografia, oito fatores devem ser levados em consideração. Eles são:
(1) a qualidade da radiografia;
(2) a relação com o seio maxilar e com o conduto dentário inferior;
(3) o número de raízes;
(4) a posição e tamanho das raízes;
(5) existência de reabsorção;
(6) se foi feito tratamento endodôntico;
(7) a perda do osso alveolar;
(8) a densidade óssea.
10.02. ATO CIRÚRGICO:
- Isolamento e desinfecção do campo cirúrgico.
- Anestesia local, infiltrativa ou regional.
- A extração dentária deve ser realizada com o mínimo de trauma possível.
- Durante a extração propriamente dita, optamos por realizá-la cirurgicamente, isto é, o procedimento inclui o afastamento dos tecidos moles, com uma sindesmotomia cuidadosa; também, a divisão do dente e das raízes, com a utilização de brocas cirúrgicas (odontosecção); e a remoção de osso alveolar para, em seguida, utilizar o fórceps ou as alavancas.
- Em pacientes hemofílicos, a extração cirúrgica é sempre indicada por ser menos traumática e pelo fato de estarmos sempre prevenidos para as possíveis surpresas. O dente é dividido de maneira controlada, facilitando, desta maneira, a sua remoção.
- O dente secionado e suas raízes são retirados com o máximo cuidado, produzindo-se assim um traumatismo mínimo.
- Seguem-se a curetagem e a remoção de tecidos infectados. Consideramos a curetagem indispensável , para eliminar os diversos fatores que podem levar a uma hemorragia, tais como: cistos, tecido de granulação, esquírolas ósseas, etc.
- A sutura é sempre indicada porque ela, por si só, se complementa como um poderoso hemostático. ( Seda preta agulhada , com agulha atraumática)
Em seguida, com o alvéolo já suturado, utilizamos o selante ou adesivo de fibrina, dentro do alvéolo. O selante de fibrina é introduzido dentro do alvéolo, pelo sistema de “duplo jet”, utilizando-se as agulhas de ponta romba que acompanham o “kit”. Utilizamos sempre o de 0,5 ml.
A ferida deve ser protegida com esponja de fibrina e comprimida com uma gaze embebida em soro fisiológico, por um período de 3 horas.
- Em dentes decíduos, via de regra após a extração de um dente decíduo não recomendamos a sutura; somente compressão local, com espoja de fibrina e trombina, bem como, compressão com gaze embebida em soro fisiológico, por um período de 3 horas.
11. CIRURGIA BUCAL MENOR
DENTES INCLUSOS OU IMPACTADOS
- O paciente deve estar previamente preparado com o fator ausente (a critério do hematologista responsável) e devidamente instruído sobre o pós-operatório, a higiene bucal, etc.
-Terceiro molar inferior incluso ou impactado: Antes de qualquer intervenção, um minucioso estudo radiográfico deve ser efetuado (Raios X panorâmico e apical).
Assim podemos visualizar a sua forma anatômica e a relação do alvéolo do terceiro molar inferior com o conduto dentário, que devem ser conhecida anatomicamente e identificada. Este é um problema que deve sempre ser considerado para evitar lesões no conduto e no seu conteúdo (artérias, veias e nervos).
- Quanto ao ato cirúrgico, devemos efetuá-lo cuidadosamente, obedecendo todos os passos das diversas técnicas cirúrgicas.
12APICETOMIAS
Não estão contraindicadas as intervenções em terço apical de incisivos e caninos, maxilar superior ou inferior, principalmente se for usado o selante de fibrina.
12.01.PÓS-OPERATÓRIO:
RECOMENDAÇÕES E MEDIDAS GERAIS:
- Todos os pacientes são atendidos em regime ambulatorial.
- As instruções do pós-operatório devem ser dadas por escrito ao paciente.
- Dieta líquida ou pastosa nas primeiras 24 horas.
- Lavar a boca com água morna (37O C) e sal, sem bochechar ou lavar a boca com um comprimido de ácido épsilon amino caproico, dissolvido em meio copo de água morna (370 C), também sem bochechar.
- Gelo por fora, durante meia hora, 4 vezes ao dia, nas primeiras 24 horas.
- Para controle da dor, paracetamol 750 mg. de 4 em 4 horas, no máximo por 12 horas ( não deve ser ministrado em caso de problema hepático).
- O ácido épsilon amino caproico, deverá ser utilizado como medicação coadjuvante na dose de 50 mg por quilo de peso, de 6 em 6 horas, durante 5 dias . Ele deve ser prescrito pelo médico que assiste o paciente, no máximo de 12 g diários e não deve ser ministrado em caso de problema hepático.
- É feita a revisão da extração no quarto dia e, finalmente, a sutura é retirada no oitavo dia pós a cirurgia, quando é dada a alta ao paciente.
CONTROLE DAS HEMORRAGIAS
- O coágulo desorganizado deve ser sempre removido, a ferida, inspecionada e diagnosticada a causa do sangramento.
- Uma vez limpa a ferida, pode-se localizar perfeitamente o local da hemorragia; esta pode se encontrar no interior do alvéolo dentário, nas partes moles ou nas suas bordas.
Um tamponamento local deve ser feito. Podemos utilizar a esponja de colágeno ou fibrina + trombina ou surgicel e /ou uma proteção com cimento cirúrgico.
- Se a hemorragia for proveniente das partes moles pode-se utilizar bolinhas de algodão embebidas em ácido tricloroacético (tendo o cuidado de retirar o excesso do ácido), para a cauterização química no local do sangramento. Pode-se, ainda, utilizar algodão embebido em perclorato de ferro.
- Em hemorragias periodontais é recomendado o uso de cyanoacrilatos.
- Nas hemorragias provocadas por acidentes, principalmente em crianças, localizadas nas partes moles e na língua, é sempre indicada a sutura, com reposição prévia de fator, utilizando-se seda preta agulhada 5.0 ou 6.0, com agulha atraumática, e sempre que possível, ser efetuada, em centro cirúrgico e sob anestesia geral.
- No caso de hemorragias recorrentes, retorna-se ao protocolo inicial.
HEMOSTÁTICOS LOCAIS
Os principais agentes hemostáticos locais, para serem colocados dentro do alvéolo dentário, após a extração são:
- Estípicos: ácido tricloroacético, percloreto de ferro, etc.
- Esponja de colágeno (Gelfoan), associada ao iodofórmio ou a pasta de Reclus.
- Esponja de colágeno e trombina bovina – somente para uso tópico.
- Placenta humana dessecada e associada à trombina bovina em um pote dapen, uma pequena tira de gaze e cimento cirúrgico. São manipulados numa placa de vidro até atingir a consistência de uma pasta; após a extração, passa-se o fio de sutura e, em seguida, o alvéolo é tamponado com esta pasta, que deve ficar um milímetro aquém do rebordo alveolar e, finalmente, è amarrado o fio de sutura.
- Esponja de fibrina (Avitene, Lyostipic).
- Celulose oxidada (Surgicel, Oxycel).
- Cyanoacrilatos (Dermabond,Indermil, Liquiband), atuam por polimerização e protegem imediatamente a ferida. Entretanto, só devem ser utilizado externamente (uso tópico), porque não são absorvidos pelo organismo.
- Selante de fibrina, ou “cola” de fibrina, ou adesivo de fibrina (Tissucol, Beriplast). Trata-se de um complexo derivado do plasma humano que imita a última fase da “cascata da coagulação”, quando o fibrinogênio se transforma em fibrina.
Em poucos dias ele é absorvido pelo organismo, dando lugar a uma rede de fibroblastos, iniciando assim o processo de cicatrização.
O selante de fibrina age como hemostático, como selante (impedindo a saída de líquidos) e, principalmente, como acelerador do processo de cicatrização. Além de ser fisiológico, o adesivo de fibrina, quando é utilizado com a trombina 4 ou lenta(Tissucol), permite a associação com bio-materiais, tais como: osso autólogo, osso de banco de osso, hidróxido de hepatita, biocerâmica, etc., para preenchimento de cavidades, notadamente aquelas em que ocorrem perdas ósseas.
Também pode ser usado com antibióticos, como por exemplo, a Gentamicina.